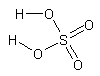
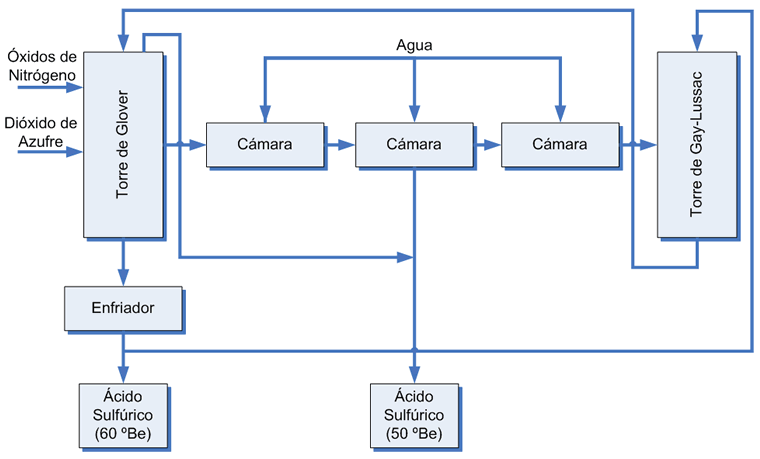
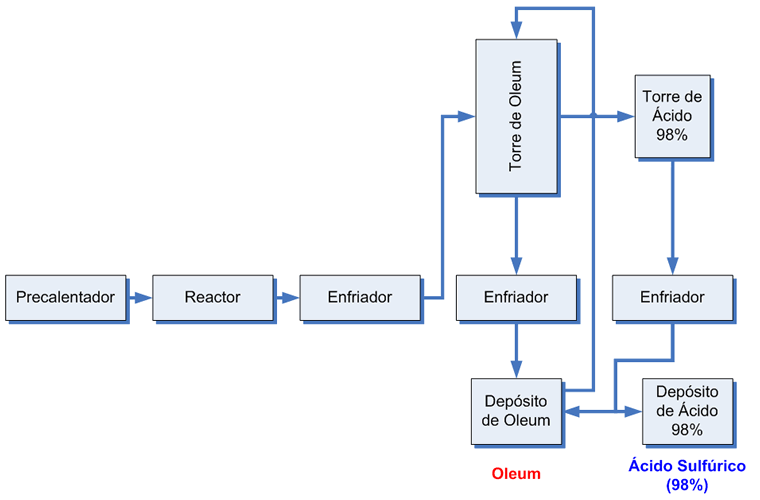
Emisiones en el proceso de contacto
Composición
La mayoría de las emisiones de las plantas
de contacto se deben al gas de cola de la torre de absorción. El gas contiene
principalmente nitrógeno y oxigeno, pero también SO
2 sin reaccionar,
SO
3 no absorbido, niebla de ácido sulfúrico y
spray de ácido.
Cuando el gas penetra en la atmósfera, el SO
3 absorbe vapor de
agua para formar una niebla ácida. Otras emisiones menores de SO
2 y
SO
3 se escapan de los tanques de almacenamientos, concentradores
de ácido y equipos de proceso.
Emisiones
de SO2
La mayor emisión de una planta de
contacto es de dióxido de azufre contenido en los gases de chimenea. El SO
2 procede
de la conversión incompleta a SO
3 en la catálisis. Se pueden alcanzar
rendimientos de conversión del 98 a 98,5 % con un diseño adecuado de planta.
Aunque es posible lograr conversiones aun más altas, estas se consiguen a
expensas del encarecimiento de la instalación y, por consiguiente, de costos
de producción más elevados.
Muchas plantas de contacto se adquieren
de fabricantes que garantizan una conversión con un rendimiento del 96 al
98%. Por regla general el diseño de las plantas prevé un rendimiento de conversión
superior al garantizado. De este modo una planta puede operar a capacidad
apreciablemente superiores a la nominal sin que el rendimiento de la conversión
disminuya.
En Alemania se patento recientemente
un proceso de contacto que reduce la concentración del SO
2 en
los gases de cola a 0,03% SO
2. El proceso consiste en la adición
al sistema de una torre de absorción de SO
3 antes de la última
etapa de conversión. La eliminación del SO
3 en este punto da lugar
a una conversión global del orden 99,7%. Este diseño no se traduce en una
elevación considerable de la inversión y los coste de producción son aproximadamente
iguales. Este proceso, de la casa Bayer, que se llama doble contacto, es
posiblemente el sistema más seguro hoy día para la reducción de las emisiones
de SO
2 en la fabricación de ácido sulfúrico.
En muchas plantas de contacto que
queman azufre, es posible reducir a 0,1% la emisión de SO
2 en
el gas de salida, operando con un gas muy diluido, aunque, como es lógico,
esto incrementa los costes de la operación.
La figura 1 muestra el porcentaje
de conversión de SO
2 a SO
3 relacionado con la concentración
de SO
2 en el gas cola para un total de 31 plantas de contacto
analizadas en los Estados Unidos. La reducción de SO
2 en la emisión
de la chimenea, esta en relación directa con el incremento de la conversión
del SO
2 en el convertidor catalítico. Las cantidades y concentración
de las emisiones de SO
2, dependen del rendimiento de la conversión,
es decir, dependen de la concentración de SO
2 en los gases de
alimentación del convertidor y relación O
2/SO
2, especialmente
en la ultima etapa de conversión, numero de etapas del convertidor catalítico,
volumen y distribución del catalizador en estas etapas, rendimiento del catalizador,
uniformidad de la composición del gas, impurezas en el gas de alimentación,
control de la temperatura en diferentes puntos del convertidor.
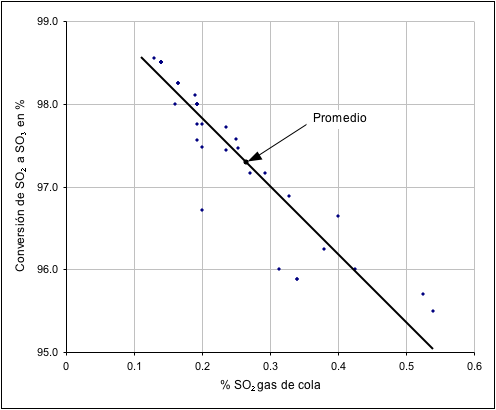 Figura
Nº 1
Figura
Nº 1 Relación del rendimiento de conversión del SO
2 en
el gas de salida.
La figura 2 resume el porcentaje
de conversión de SO
2 a SO
3 en las plantas que queman
azufre sin aire de dilución y la figura 3 el de las que queman azufre con
aire de dilución. En la figura 4 se presenta las emisiones de SO
2 para
varios rendimientos de conversión de SO
2.
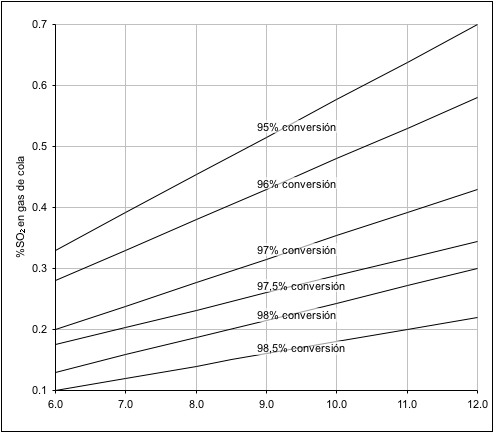 Figura
Nº 2
Figura
Nº 2 Porcentaje de conversión del SO
2 a
SO
3 para plantas sin dilución con aire.
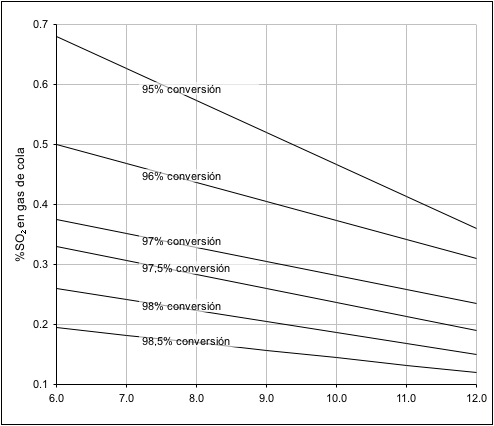 Figura
Nº 3
Figura
Nº 3 Porcentaje de conversión de SO
2 a
SO
3 para plantas con dilución, con aire.
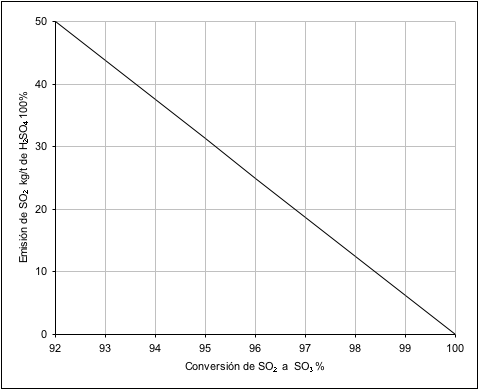 Figura
Nº 4
Figura
Nº 4 Emisiones de SO
2 para
diferentes rendimientos de conversión (por
t de sulfúrico % producido).
Emisiones
de niebla ácida y spray
Los datos recogidos en 33 plantas
de contacto norteamericanas indican que la niebla ácida y el spray ácido
que contienen los gases de cola varían entre 380 y 1720 mg/Nm
3.
El valor medio de los datos es 445 mg/Nm
3.
Existen tres tipos diferentes de
recuperación de niebla y spray ácidos: precipitadores electrostáticos, filtros
de fibra y vidrio y filtros de tela metálica. En 14 análisis de 10 planta
en diferentes condiciones, que contaban valores de 63 a 826 mg/Nm
3.
La tabla Nº 1 refleja el efecto de los filtros de tela metálica en un sistema
de dos etapas para una unidad que opera con una velocidad de gas 3,5 a 4,5
m/s
Emisiones
de SO3
Al entrar en contacto con la humedad
de la atmósfera el SO
3 no absorbido se hidrata a ácido sulfúrico
y forma una pluma blanca visible de niebla ácida.
Los valores tomados en algunas planta
norteamericanas indican que la emisión varía desde menos de 15 mg/Nm
3 hasta
1700 mg/Nm
3 de SO
3. Por regla general, la formación
de pluma visible se debe a condiciones de funcionamiento deficiente de la
planta, que pueden corregirse con un mantenimiento adecuado.
Tabla Nº1
Efecto de los separadores de niebla
de malla metálica sobre la recogida en niebla ácida
|
Caída
de presión,
mm de agua
|
Niebla ácida
mg/m3 de
gas de cola
|
Rendimiento de separación de
niebla ácida,
%
|
|
Entrada
|
Salida
|
|
19
|
146,4
|
26,4
|
92,0
|
|
25,4
|
314,1
|
24,9
|
92,5
|
|
31,7
|
492,6
|
32,8
|
93,4
|
|
31,7
|
2388,3
|
27,5
|
98,9
|
|
38,1
|
1160,3
|
35,0
|
97,0
|
|
38,1
|
464,1
|
27,1
|
94,1
|
|
Media
|
828,2
|
29,0
|
96,5
|
Emisiones
en las paradas y puestas en marcha de la planta
En las plantas de tostación de piritas
una parada repentina puede producir una elevada emisión de SO
2,
generada por la evacuación de los gases de tostación a la atmósfera.
Según las características del clima,
del tamaño de la pirita y aislamiento, las paradas programadas pueden variar
entre 8 a 24 horas, sin que se registre un excesivo enfriamiento n de la
cámara catalítica ni del resto del equipo. Si la duración de la parada fue
muy larga es preciso precalentar el catalizador con un equipo auxiliar.
Cuando la temperatura de absorción
desciende por debajo de los 65ºC el rendimiento de absorción decrece y la
emisión de SO
3 y niebla ácida es muy superior a la normal.
Las perdidas de SO
2 y
niebla ácida durante la puesta en marcha subsiguiente a una parada de larga
duración pueden ser muy grandes si es que el equipo no se ha precalentado
convenientemente. La duración de estas perdidas anormales puede durar desde
varios minutos hasta 12-19 horas, según sea el régimen de temperaturas alcanzado
y del ajuste a las condiciones de operación de todo el equipo.
Métodos de reducción de las emisiones en el
proceso de contacto
Como ya se vio, la mayor emisión de una planta
de ácido sulfúrico por contacto corresponde al SO
2. La cantidad
y concentración del SO
2 en los gases de cola depende principalmente
del rendimiento de la conversión en SO
3.
El primer procedimiento para aumentar
la conversión consiste en el cambio periódico de las masas catalíticas a
fin de incrementar el rendimiento en la catálisis. Aunque es el procedimiento
más viable para reducir la emisión de SO
2, Solo en muy pocos casos
se logra alcanzar el limite máximo de conversión y aun en estos no podría
mantenerse.
Hay que tener en cuenta que aparte
del coste propio del catalizador el cambio de la masa catalítica acarrea
gastos adicionales notables, pues exige parar la planta, descebar el convertidor,
cambiar las masas, poner el convertidor en régimen de temperaturas y, a continuación,
volver a arrancar. El tiempo que suele invertirse en estos cambios de masa
determina que esta operación solo se efectúe cuando el rendimiento de la
catálisis haya bajado mucho al cabo de periodos de funcionamiento prolongados.
El segundo procedimiento puede consistir
en la transformación de una planta de catálisis simple en otra de doble catálisis,
pero aun cuando el rendimiento mejoraría considerablemente esta operación
es económicamente prohibitiva dados los cambios, transformaciones y ampliaciones
de equipo a introducir en la planta. Por lo tanto no es una solución factible
desde un punto de vista exclusivamente económico.
El tercer procedimiento consiste
en el empleo de chimeneas altas. La altura media de las chimeneas de las
plantas de ácido sulfúrico es de 12 a 30 m aunque también existe un cierto
número de chimeneas de 120 m y más.
Se puede aumentar la altura efectiva
de una chimenea recurriendo a la acción de aire caliente o gases residuales
de otro proceso de combustión. Las partículas de ácido pueden eliminarse
con una chimenea de gran altura o de mayor diámetro, que disminuya la velocidad
del gas en la chimenea, y les permita depositarse en las paredes.
Los resultados analíticos de una
serie de ensayos realizados en una planta norteamericana con una chimenea
de 75 m de altura, demostraron que la concentración de niebla ácida y spray
iba desde 900 mg/Nm
3, en la base de la chimenea, hasta 80 mg/Nm
3 en
la cabeza. De este modo se recupera en la chimenea el 91% del peso total
de niebla ácida y spray.
De los estudios teóricos realizados
sobre la distribución de la contaminación por difusión, se observa que la
concentración del contaminante en los alrededores varia en razón inversa
al cuadrado de la altura de la chimenea. Esto quiere decir que si la altura
de la chimenea se dobla, la emisión se reduce en una cuarta parte.
Un cuarto procedimiento consiste
en la adopción de procesos de desulfuración de los gases de cola. En la tabla
Nº 2 se resumen los procesos de desulfuración que actualmente se estudian
y ensayan en el mundo.
El análisis y estudio de la información
disponible sobre esos procesos lleva a las conclusiones siguientes:
La mayoría aun
no ha alcanzado la fase de explotación industrial.
Como todos se
orientan preferentemente a la desulfuración de los gases de cola en centrales
térmicas.
Como deben tratar
un gran volumen de gases para reducir su costo de explotación, son prácticamente
inadaptables a plantas de ácido sulfúrico.
Surgen dificultades
adicionales cuando se intenta aplicarlos a plantas ya existentes.
Por las razones
anteriores, y sin entrar en su evolución económica, se considera que hoy
en día aun no puede pensarse en la desulfuración de los gases de cola de
las plantas de ácido sulfúrico.
TABLA Nº 2
PROCESOS PARA LA ELIMINACIÓN DE SO2
| Proceso |
Fundamento |
Subproducto |
| Basterra
and Bankside |
Lavado con
aguas del río Támesis |
Ninguno |
| Wisconsin
Electric Company |
Lavado con
agua de régimen turbulento |
Ninguno |
| Combustion
Engieenering |
Inyección
de dolomita y lavado en húmedo |
Ninguno |
| Mitsubishi |
Absorción
de SO2 con MnO2 |
Sulfato
amónico |
| Showa
Denko |
Lavado con
amoniaco |
Sulfato
amónico |
| U.S.
Bureau of Mines |
Absorción
de alúmina alcalinizada |
Azufre
elemental |
| Still |
Absorción
con cenizas de linguitos |
SO2 para
planta de ácido |
| Grillo |
Absorción
con óxidos e hidróxidos alcalinos |
SO2 para
planta de ácido |
| Monsanto-Penelec |
Oxidación
catalítica con V2O5 |
H2SO4 80% |
| Kiyoura
T.I.T. |
Oxidación
catalítica con V2O5 e inyección de amoniaco |
Sulfato
amónico de elevada pureza |
| Sulfacid
(Lurgi) |
Catálisis
húmeda sobre carbón activo |
H2SO4 (50-75%) |
| Hitachi |
Catálisis
húmeda sobre carbón activo |
H2SO4 (15-25%) |
| Reinluft |
Absorción
sobre lechos de carbón activo |
SO2 para
planta de ácido |
| Carbonato
fundido |
Absorción
de carbonatos alcalinos fundidos |
Azufre
elemental |
| Checoslovaco |
Absorción
con amoniaco de descomposición con ácido nítrico |
SO2 y
nitrato amónico para planta fertilizante |
Concentraciones de emisión producidas
Se termina con el análisis de los resultados
obtenidos en el estudio experimental de una planta de ácido sulfúrico egipcia.
Los datos de operación de la planta
se reflejan en la tabla Nº 3, en la que los resultados se obtuvieron dividiendo
la suma de los valores registrados en periodos de varios días por el número
de días de cada uno de estos periodos.
Se realizaron dos tipos de determinaciones
de inmisión de SO
2: mediante el método del peróxido de plomo se
realizaron análisis en puntos alejados de la chimenea (en un radio de 1 Km.
aproximadamente) y mediante valoración por iodimetria se realizaron los análisis
en la misma planta (en un radio de 15 m) tomando como centro la base de la
chimenea de 2 metros.
Los resultados obtenidos por iodimetria
dieron un margen de concentración de SO
2 de 32 a 11 ppm siendo
el valor medio en el ambiente de la planta 5 ppm.
En las curvas de disolución media,
calculada a partir de los datos de análisis obtenidos en el total del periodo,
por el método del pentóxido de plomo, se acusan además los efectos de la
velocidad y dirección del viento. El efecto de la velocidad queda algo mas
enmascarado por el posible efecto de los edificios de la factoría, así como
por otras fuentes de las inmediaciones.
En este caso particular, el valor
de 5 ppm de SO
2 en la atmósfera de la planta es muy alto en comparación
con las concentraciones de las plantas térmicas e instalaciones siderúrgicas
estudiadas de esta misma forma que no exceden las 0,3 ppm de SO
2 en
régimen normal.
La concentración de 5 ppm puede tener
efectos serios para la salud de las personas que trabajan en los alrededores
de la planta de ácido.
La solución que se recomendaba en
el estudio era aumentar la altura de la chimenea a 40 metros y la instalación
de una torre de lavado (scrubber) de los gases con agua y de un precipitado
de niebla ácida antes de expulsar el afluente a la atmósfera.
Inversiones y costos
Si, por ejemplo, se trata de una
planta ya instalada de 1000 tm/día con una emisión de 13 t SO
2/día,
la instalación de un sistema de depuración con piridina para obtener sulfato
amónico (producto de difícil salida), exigirá una inversión de 43 x 10
2 dólares.
En los Estados Unidos la planta de
la Olin Corp. en Taulsboro de una capacidad de producción de 257.000 t/año
emplea el sistema de lavado de los gases de cola con sulfito-bisulfito de
sodio desarrollado por la Wellman Power Gas Co. Pero para la adopción de
este proceso, la planta debe ubicarse en las proximidades de una fábrica
papelera con el objeto de que absorba las grandes cantidades de sulfito de
sodio necesarias.
Los valores limites señalados recientemente
por la EPA (Envioronmental Protection Agency) de los estados unidos son de
480 ppm para el SO
2 en los gases de cola, aunque los usuales en
la mayoría de los países son de 2000 ppm de SO
2.
En términos de inversiones, la EPA
ha calculado que la inversión en una planta de 250.000 t/año de ácido sulfúrico
que carezca de equipo antipolución asciende a 3 x 10
6 dólares.
La instalación de equipo diseñado para mantener la polución por debajo de
las 480 ppm de SO
2 costara unos 900.000 dólares más; ello representa
un aumento del 30%. Por otra parte los costos de fabricación se incrementan
alrededor de un 5%, es decir, en unos 31,5 $/t.
En el montaje de una nueve planta,
pueden servir como cifras indicativas las inversiones realizadas en una de
contacto simple y en otra Bayer de doble absorción con y sin sistema de depuración
convencional.
Para una capacidad de 700 t/día la
inversión en la planta de contacto simple es de 415 millones de pesetas,
en la de doble absorción 460 millones (13 % más) y con sistema de depuración
de 2 millones de dólares, es decir, un 2% mas. Todos estos datos se refieren
a una emisión límite de 2000 ppm.
1. Generalidades.
Mientras
que todos los vertidos urbanos presentan impurezas minerales y orgánicas
cuya naturaleza y concentración son bastantes similares de una ciudad a otra,
y por ello sus líneas de tratamiento son análogas, los vertidos industriales,
debido a su gran diversidad, necesitan una investigación propia de cada tipo
de industria y la aplicación de procesos de tratamientos específicos.
Pueden citarse algunos factores principales
que la contaminación industrial tiene en común con la contaminación de origen
urbano, pero las vías de depuración, normalmente, deben definirse industria
por industria.
Al enumerar las principales industrias,
se ve que según las contaminaciones que producen, justifican tratamientos
biológicos (parecidos a los de las aguas urbanas) o tratamientos estrictamente
químicos (como en las industrias de ácidos)
Las estaciones de tratamiento de
aguas industriales se destinan a cumplir unas normas de vertido, que no se
refieren únicamente a la de
D.B.O., a la
D.Q.O. y a
los contenidos de
materiales en suspensión, sino, también, a un cierto
número de compuestos minerales y orgánicos. Por otra parte, estas normas
se definen, en varios países, según las diversas ramas profesionales.
La definición de todo tratamiento
deberá basarse en:
- el conocimiento de los diversos contaminantes;
- la caracterización de los efluentes;
- la organización de los desagües y la
separación de los efluentes;
La elección entre los diversos métodos de
depuración fisicoquímica y/o biológica.
Por lo tanto, el buen funcionamiento
de la instalación dependerá de que se realice previamente un estudio minucioso,
ya que cualquier elemento nocivo, que no se hubiera tenido en cuenta, podría
perturbar seriamente la instalación.
2. Factores específicos de contaminación.
Las
aportaciones significativas de contaminación que se enumeran seguidamente,
se han clasificado en función de los métodos de tratamiento que le son aplicables.
- Elementos insolubles
separables físicamente con o sin floculación: materias grasas, flotantes
(grasas, hidrocarburos alifáticos, alquitranes, aceites orgánicos, etc.).
Materias
sólidas en suspención (arenas, óxidos,
hidróxidos, pigmentos, azufre coloidal, látex, fibras, etc.).
- Elementos orgánicos
separables por adsorción: colorantes, detergentes, compuestos macromoleculares
diversos, compuestos fenolados.
- Elementos separables
por precipitación: metales tóxicos o no, Fe, Cu, Zn, Ni, Be, Ti, Al,
Pb, Hg, Cr, precipitables en una cierta zona de PH; sulfitos, fosfatos,
sulfatos, fluoruros, por adición de Ca2+.
- Elementos que pueden
precipitar en forma de sales insolubles de hierro o de complejos: sulfuros,
fosfatos, cianuros, sulfocianuros.
- Elementos separables
por desgasificación o stripping: H2S, NH4,
alcoholes, fenoles y sulfuros.
- Elementos que necesitan
una reacción de oxidación-reducción: cianuros, cromo hexavalente, sulfuros,
cromo, nitrito.
- Ácidos y bases: ácido
clorhídrico, nítrico, sulfúrico y fluorhídrico; bases diversas.
- Elementos que pueden
concentrarse por intercambio iónico o por ósmosis inversa: radionucleidos
tales como iodo, Mo, Cs; sales de ácidos y de bases fuertes; compuestos
orgánicos ionizados (intercambio iónico) o no (osmosis inversa).
- Elementos que se
adaptan a un tratamiento biológico: todos los elementos biodegradables
por definición; por ejemplo, azúcares, proteínas, fenoles. Los tratamientos
biológicos pueden aplicarse también, después de su aclimatación, a compuestos
orgánicos tales como el formol, la anilina y ciertos detergentes.
Deben recordarse los puntos siguientes:
- La relación entre la D.Q.O.
y la D.B.O. en aguas industriales es muy diferente de la que se obtiene en
aguas domésticas. Esta relación evoluciona en las diversas fases del tratamiento,
pudiendo llegar la D.Q.O. final, en algunos casos, hasta más de 5 veces
el valor de la D.B.O. correspondiente.
- La presencia de tóxicos
muy activos puede enmascarar la presencia de materias biodegradables
y falsear la medida de la D.B.O..
- Nociones de tratabilidad
biológica de los efluentes.
3. Caracterización de los efluentes.
Para
la buena definición de una estación de tratamiento de aguas residuales, es
necesario disponer de los siguientes datos, cuidadosamente establecidos:
- Volúmenes diarios;
- Caudales horarios mínimo y máximo;
- Composición del agua de aportación a la
fábrica;
- Fabricaciones continuas, discontinuas;
- Importancia y periodicidad de las puntas de
contaminación;
- Posibilidad de separación de circuitos;
- Posibilidades de tratamientos o de recirculaciones
locales o parciales;
- Contaminaciones secundarias, incluso débiles
u ocasionales, que puedan afectar seriamente al funcionamiento de ciertos órganos
de los equipos de tratamiento (colas, alquitranes, fibras, aceites, arenas,
etc.).
Al realizar el proyecto de una fábrica,
estos datos, recogidos después del análisis de las fabricaciones, deben compararse
con informaciones procedentes de fábricas existentes.
Cuando se trata de acondicionar una
fábrica ya existente, conviene realizar una comparación de las cantidades
de contaminantes, detectados mediante un análisis continuo y sistemático
de los efluentes, con los consumos de productos químicos de la fábrica.
4. Tratamientos separados.
A veces resulta conveniente aislar ciertos efluentes
y someterlos a un tratamiento específico. Se impone esta forma de proceder
siempre que el efluente que procede de una unidad de fabricación presenta
una de las siguientes características:
- Concentraciones muy elevadas de D.Q.O. o de
D.B.O. debido a la presencia de compuestos solubles;
- Concentraciones medias o elevadas de H2S,
de NH4 o de elementos tóxicos.
En lugar de diluir estos efluentes,
suele ser más económico utilizar uno de los siguientes procedimientos:
- Concentración con vistas a reutilizar el producto;
- Destrucción por pirólisis directa del líquido
o del vapor procedente de su stripping;
- Extracción líquido-líquido.
Se citan tres ejemplos de reducción
de la contaminación del efluente general de una fábrica:
- Regeneración de baños usados muy diversos (galvanoplastita,
mecánica), por eliminación discontinua o continua de sus impurezas en disolución
o en suspensión;
- Tratamiento químico de licores de sales o de ácidos
cuya concentración es superior al umbral de solubilidad de la sal de calcio
correspondiente, que puede entonces precipitar;
- Tratamiento de aceites solubles por vía química,
térmica o por separación por membranas.
5. Tratamientos preliminares.
Las condiciones de tratamiento previo de los
efluentes generales de fábricas son también más variadas que en el caso de
aguas residuales urbanas.
Las operaciones de
desbaste automático
son deseables en la mayoría de las industrias e indispensables en algunas
de ellas.
El
desarenado sólo se realiza
en algunos casos particulares; y el
desaceitado se utiliza con bastante
frecuencia: los hidrocarburos y aceites proceden a veces de fabricaciones,
y sistemáticamente de los circuitos de engrase o de almacenamiento de carburante.
También se prevé frecuentemente la
regulación del
caudal hidráulico y de la carga contaminante, que puede llevarse a cabo:
- Mediante el empleo
de depósitos derivados, en los que se almacena el agua de tormentas
cuando la red es unitaria y cuando las lluvias, de volumen siempre menor
que en áreas urbanas, arrastran y diluyen contaminantes. La finalidad de
estos depósitos es, por lo tanto, la de evitar que la línea de tratamiento
haya de dimensionarse en función de unas puntas excepcionales de caudal.
- Mediante el empleo
de depósitos de homogeneización, en los que se almacena durante
algunas horas, e incluso por espacio de varios días, la totalidad de los
efluentes producidos por una unidad o por toda la fábrica. Es indispensable
prever la agitación de estos depósitos. Su objetivo es el de reducir las
puntadas de contaminación, con el fin de evitar sobrecargas de concentración
perjudiciales para el funcionamiento regular de la línea de depuración. Con
ello, se consigue, a demás, un cierto grado de previsión en la explotación.
Algunas veces se realizan operaciones
previas de neutralización, de oxidación y de reducción, para tratar efluentes
concentrados o tóxicos. En estas operaciones intervienen autómatas de regulación
de PH o de potencial redox.
6. Tratamientos fisicoquímicos.
La
depuración fisicoquímica puede constituir, según el caso, una etapa intermedia
o la etapa final del tratamiento total. Tiene una o varias finalidades:
- Precipitación de metales o de sales tóxicas;
- Eliminación de aceites en emulsión y de materias
diversas en suspensión;
- Clarificación con reducción simultánea de D.B.O.
coloidal y de la D.Q.O. correspondiente.
Este tratamiento implica la necesidad
de mantener una zona de PH bastante reducida, y, según la naturaleza del
proceso (precipitación, cristalización, adsorción o floculación), puede realizarse
en reactores-decantadores o clarificadores muy diferentes:
- Flotadores tales como el Flotazur o
el Sediflotazur, en eliminación de aceites o fibras;
- Reactores, como el Turbactor,
y reactores-decantadores, como el Circulator, el Densator o
el Turbocirculator, para la precipitación de sales o hidróxidos;
- Clarificadores-reactores de circulación
de fangos, como el Turbocirculator, el decantador R.P.S.,
el Accelator,
en situaciones mixtas;
- Clarificadores de lechos de fangos,
como el Pulsator y el superpulsator, cuando es preciso
separar un flóculo tenue o cuando se desea desarrollar las propiedades
adsorbentes del lecho de fangos.
La elección entre estos aparatos
depende no sólo del proceso dominante que haya de efectuarse, sino también
de otros parámetros propios de la industria considerada.
Según las circunstancias, esta depuración
fisicoquímica puede ir precedida o seguida de uno de los procesos siguientes:
- Neutralización;
- Oxidación
o reducción;
- Desgasificación
o stripping.
Sólo se requiere una filtración en
caso de normas de vertido muy estrictas relativas a la materia en suspención
y a los metales totales.
7. Tratamientos biológicos.
La
posibilidad de recurrir a tratamientos biológicos, es decir, a una depuración
de tipo biológica depende de la biodegradabilidad de los efluentes, y deben
tenerse en cuenta, en su concepción, ciertas peculiaridades de las aguas
industriales:
Las aguas que se han sometido, generalmente
con varios propósitos, a un tratamiento fisicoquímico previo, se encuentran
poco cargadas de materias en suspención;
Su composición en nutrientes casi
nunca es equilibrada, con lo que debe practicarse una corrección referente
al fósforo y/o al nitrógeno;
Una deficiencia inicial de microorganismos
debe compensarse mediante una siembra adecuada y la aclimatación de organismos
específicos;
La presencia de compuestos biodegradables
puede hacer necesario el mantenimiento de una relativa constancia de su concentración
y el desarrollo de una flora específica;
Las concentraciones demasiado elevadas
de sales minerales y, sobre todo, sus variaciones rápidas, pueden perturbar
el desarrollo de la depuración;
La nitrificación-desnitrificación
puede verse afectada por unas concentraciones demasiado elevadas de D.Q.O.
y de amoníaco y dentro de ciertas zonas de PH;
Debe prestarse una atención especial
al mantenimiento de zonas de temperaturas bastante constante, ya que la temperatura
de ciertos efluentes favorece el desarrollo de bacterias termófilas.
Composición de la línea de tratamiento:
Fangos activados
de alta carga, a media carga o, en el caso general, en aeración prolongada;
Lechos bacterianos
de materiales plásticos ordenados, en pretratamiento o en tratamiento de
afino;
Lechos bacterianos
tradicionales;
Filtros de
biolite,
en tratamiento principal o en tratamiento de afino;
Lagunas aireadas
o mixtas, en tratamiento de afino.
8. Eliminación de la D.Q.O. no biodegradable.
La
depuración biológica constituye la vía más racional para la reducción de
la D.B.O. y de su correspondiente D.Q.O.. Sin embargo, la aplicación de reglamentaciones
cada vez más rigurosas puede hacer que sea necesaria la eliminación complementaria
de la D.Q.O. no biodegradable, de color y de ciertos compuestos específicos.
Esta D.Q.O. se debe a compuestos
orgánicos, en general disueltos y de naturaleza muy diversa: disolventes,
hidrocarburos aromáticos, derivados nitrados y sulfonados, etc...
Los procedimientos usuales para la
eliminación de esta D.Q.O. son los siguientes:
Primero: adsorción a través de carbón
activo con regeneración térmica o química o a través de adsorbentes diversos.
Segundo: ultrafiltración y ósmosis inversa.
Tercero: intercambio iónico.
Cuarto: oxidaciones diversas (aire, oxígeno,
ozono, cloro).
9. Fangos industriales.
Naturalmente,
el carácter específico de las aguas residuales industriales se observa también
en los fangos producidos, que a veces son de predominio y otras -y este es
el caso más frecuente- de predominio mineral.
En general, los fangos de depuraciones
fisicoquímicas son más abundantes que los que proceden de las depuraciones
biológicas. Se observa, por último, que los fangos procedentes de la clarificación
de aguas de aportación industriales son a veces preponderantes. Todas la
técnicas de tratamiento que se han definido para los fangos urbanos son igualmente
aplicables en este caso. Se citan simplemente algunas particularidades relativas
al espesamiento y a la deshidratación mecánica.
A. Espesamiento de fangos:
El espesamiento
se lleva a cabo, sobre todo por decantación, aplicándose cargas superficiales
muy variables, de 10 a 800 Kg. MS/(m2.d), según la composición de los fangos.
La presencia de hidrocarburos en cantidad apreciable puede dar lugar a una
segunda fase líquida en el espesador y dificultar se funcionamiento.
B. Deshidratación de fangos:
El
volumen de los fangos orgánicos producidos no justifica, en general, el empleo
del acondicionamiento térmico. Por el contrario, es frecuente la realización
de un acondicionamiento químico, utilizándose polielectrolitos sintéticos
y/o reactivos minerales, y, en menor escala, cargas de materias inertes.
Los condicionamientos de filtrabilidad
o de sedimentabilidad por escurrido centrífugo de los fangos difieren no
sólo según su composición química, sino también según su modo de formación.
Se producen variaciones de filtrabilidad en las relaciones de 1 a 10, según
los productos, y de 1 a 3 para un mismo producto.
Cuando no se conocen exactamente
las características de los fangos, es indispensable realizar ensayos previos.
Para mejorar la filtrabilidad de
un fango, puede recurrirse, a veces a ciertos subproductos de la fábrica.
C. Destino final de los fangos:
Según la naturaleza
de los fangos, la formas de evacuación de los mismos son muy diferentes:
Los fangos minerales
relativamente estables y no tóxicos, pueden esparcirse como abono, descargarse
en escombreras al aire libre o utilizarse como material inerte.
Los fangos minerales
inestables o tóxicos deben almacenarse en vertederos controlados, estabilizarse
o incluso; en algunos casos, tratarse por incineración; los fangos tóxicos,
y en especial los que contengan metales pesados, sólo podrán almacenarse
en vertederos estancos, aislados de toda capa freática.
Los fangos orgánicos,
fermentables en general, deben estabilizase antes de su esparcimiento descarga
en vertedero, o eliminarse por incineración.
Cuando los fangos
ricos en aceites no son recuperables, deben incinerarse. Cuando son muy autocombustibles,
pueden contribuir a la eliminación, por incineración, de otros fangos más
pobres.
Plantas fabricadoras
de ácidos.
Tratamientos.
Los desechos de la fabricación de ácidos,
son extremadamente contaminantes, por lo cual deben tratarse antes de poderse
arrojar en los cursos de aguas. Varios son los puntos importantes, sin embargo,
el pH de estos residuos es lo que más atentamente se debe controlar y se
debe ver que dicho valor se encuentre en el rango de 6.0 a 9.0.
El principal método de tratamiento
que se efectúa a estos efluentes es el de neutralización.
Gehm nos
describe su método de neutralización de desechos ácidos, que consta de una
cama de piedra caliza, la cual trata a los desechos por encima 10.000 p.p.m.
de acidez mineral en una cama capaz de recibir 0.1 mgd de desechos. Por
otra parte también encontramos el método de
Shugart; un proceso
que utiliza caliza para una neutralización automática.
Métodos de Spray-burning
y Combustión indirecta:
- Spray-burning: Este método
consiste en esparcir el desecho ácido en una cámara de combustión a alta
temperatura (1700-2000 °F) con pequeñas cantidades de aire en exceso con
el fin de oxidar hidrocarburos. El sulfato se transforma en SO2 y
los hidrocarburos en CO2 y H2O; los gases son enfriados
y secados, y el SO2 es recuperable para su posterior uso en
la fabricación de ácido sulfúrico nuevamente.
- Combustión indirecta: La
principal reacción de éste método, es la reducción de los excedentes de ácido
sulfúrico en el sedimento fangoso por ciertos hidrocarburos que pueden
hallarse presente o que se pueden adicionar.
Método de cama de piedra
caliza :
Este
método es aplicable al caso de que una planta no sólo produzca ácido sulfúrico,
sino también otros como el clorhídrico nítrico, etc..
Consiste en una cama de piedra caliza
de neutralización ácida, unidad que se emplea en la neutralización de una
mezcla de sulfúrico y clorhídrico (comúnmente) en distintas concentraciones.
Los desechos deben diluirse hasta que alcancen una concentración de un uno
por ciento y luego se los hace pasar a través de dicha cama de piedra caliza
hacia arriba por sus tres pies de extensión en cantidades de 20 a 30 galones
por minuto por pie cuadrado del área de la cama. El inconveniente es que
se debe tener inicialmente un pH que ronde los valores 4,6 a 4,9.
Estos
tres últimos métodos son empleados principalmente por empresas
que no sólo producen ácido sino también hidrocarburos,
sales, etc. Son métodos muy generales.
Proceso de neutralización.
Introducción.
Un desecho
industrial con una alta cantidad de contenido de base o ácido, no debería
ser descargado a la corriente de agua sin un tratamiento previo. La corriente,
aun siendo de baja clasificación (llamamos de baja clasificación a una corriente
destinada a recibir desechos y/o para la navegación), es adversamente afectada
por valores altos o bajos de pH. Esta condición adversa es todavía más critica
cuando repentinamente corrientes de desechos ácidos son impuestos sobre la
corriente de agua.
Los tratamientos de los que hablamos
anteriormente, son procesos de neutralización, y entre éstos hallamos un
alto número de métodos para eliminar el alto contenido de sobreacidez en
los efluentes industriales; como por ejemplo:
- mezcla de desechos
de modo que el efecto en red de finalmente un pH neutro;
- tratamientos
mediante lechos de piedra caliza;
- mezcla de desechos
con trozos de piedra caliza o piedra dolomítica;
- el agregado
de las correctas proporciones de soluciones concentradas de soda cáustica
(NaOH) o ceniza sodada (Na2CO3) a los desechos ácidos.
Los materiales y los métodos deben
ser seleccionados en base al costo total de la operación, ya que los costos
del material varían ampliamente y los equipos que utilizan varios agentes
pueden diferir con el método seleccionado. El volumen, tipo y la cantidad
del desecho ácido a ser neutralizado son también factores al decidir qué agente
de neutralización se empleará.
En una línea de neutralización, el
ingeniero responsable debe de establecer un mínimo aceptable para el valor
de pH del efluente y adecuar el tiempo de reacción para el efluente ácido
para llegar a dicho valor. Aunque este trabajo previo se considere muchas
veces innecesario y costoso. En muchos casos, una fábrica puede reducir los
costos de la neutralización mediante la provisión de suficiente tiempo de
retención y sacrificando el nivel de eficiencia en el subsecuente tratamiento
biológico. Para el tratamiento biológico es siempre conveniente que el valor
de pH este cercano a la neutralidad, la neutralización previa le da una mayor
eficiente.
Métodos de neutralización.
1°)
Mezcla de desechos básicos y ácidos: La mezcla de efluentes puede ser
llevada a cabo en una planta simple de operación o entre fábrica vecinas.
Desechos ácidos y básicos pueden ser producidos individualmente dentro
de una planta y producirse la mezcla de los líquidos en un tiempo apropiado
lo que nos permitiría llevar a cabo la neutralización, sin embargo esto
requiere usualmente un depósito de cada efluente para abolir corrientes
excesivas de uno u otro.
Si una planta produce desechos tóxicos
alcalinos, los cuales pueden ser transportados a una planta vecina que produzca ácidos,
un económico y fehaciente proceso de neutralización nacerá de ambas. Por
ejemplo, una planta de materiales de construcción produce un efluente básico,
luego de una ecualización, de alrededor una mitad de mil para mezclar con
un efluente ácido de una química, se puede obtener una neutralización total
de ambos efluentes; siendo de bajo costo, y evitándose problemas de política
y de ingeniería. En otra instancia, se reportó el uso de 500000 galones en
reserva, de un digestivo anaerobio para mezclar desechos de planta anteriores
al tratamiento. El resultado del valor del pH de la reserva oscilará en rangos
de 6.5-8.5.
2°)
Tratamiento con piedra caliza: El pasar un efluente ácido a través
de lechos de piedra caliza, fue uno de los métodos tradicionales para la
neutralización. Los líquidos pueden ser pasados de arriba hacia abajo o
viceversa, dependiendo de el origen del aparato y del costo inherente.
Se logra escurrir un galón por minuto (gpm) por pie cuadrado o menos. La
neutralización procede químicamente acorde a la siguiente reacción:
CaCO
3 +
H
2SO
4 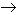
CaSO
4 +
CO
2 +
H
2O
La reacción continuará mientras dure
la disponibilidad de la piedra caliza a la misma, y en estado activo. La
primera condición puede encontrarse simplemente mediante la provisión de
cantidad suficiente de caliza; la segunda condición es aveces más difícil
de mantener. Si se trabaja con una solución de ácido sulfúrico, debe ser
diluida a un límite máximo de un 5 % y admisible hasta un rango de 5 gpm/ft
2 antes
de hacerla pasar por el lecho. No se debe concentrar los esfuerzos en neutralizar
el ácido sulfúrico arriba de un 0,3 % de concentración o a un rango menor
a 1gpm/ft
2 debido a la baja solubilidad del sulfato de calcio.
Un exceso de ácido va a precipitar el sulfato de calcio, causando subsecuentemente
una capa que pasivará la piedra caliza.
El uso del lecho de piedra caliza
puede ser una seria desventaja para este método de neutralización, ya que
la piedra caliza gastada debe ser reemplazada por nueva a intervalos periódicos,
la frecuencia del reemplazo dependerá de la cantidad y calidad de los desechos ácidos
que son pasados a través del lecho. Cuando se produce la existencia extrema
de cargas de alta acidez, se puede producir espuma, especialmente cuando
se encuetra presente material orgánico en el líquido.
3°)
Tratamiento Lime-Slurry: Mezclar desechos ácidos con una mezcla acuosa
de cal es un procedimiento efectivo de neutralización. La reacción es similar
a la del método de lechos calizos. En este caso, sin embargo, la cal es
utilizada constantemente porque es convertida en sulfato de calcio y eliminada
con el efluente. Si bien actúa lentamente, la cal posee un alto poder neutralizante
y su acción puede ser acelerada por calentamiento u oxigenación de la mezcla.
Es un proceso relativamente barato, pero a gran escala el valor puede pasar
a ser un ítem importante.
La cal hidratada es a veces difícil
de manejar, ya que tiene una tendencia a arquearse, o puentearse, sobre la
salida del depósito y posee pobres propiedades de fluidez, pero es particularmente
adaptable a los problemas de la neutralización envolviendo pequeñas cantidades
de desecho, mientras éste puede ser depositado en embalajes con la ventaja
de no tener que construir depósitos especiales.
En casos actuales, la neutralización
del ácido sulfúrico en concentraciones por encima de un 1.5%, fue llevada
a cabo satisfactoriamente por el uso de piedra dolomítica al rojo (muy alta
temperatura) que contiene 47.5% de óxido de calcio, 34.3% de óxido de magnesio
y un 1.8% de carbonato de calcio. La concentración de ácido fue limitada
hasta el estado de un 1.5%, debido a la ausencia de agua para diluir y así variar
los porcentajes. Esta cal provee la ventaja adicional de retener el sulfato
residual hasta un mínimo, una imposibilidad con cualquiera de los métodos
de lechos cálcicos.
4°)
Tratamiento de la soda cáustica: El agregado de soluciones concentradas
de hidróxido de sodio a los desechos ácidos como así también el agregado
de carbonato de calcio en concentraciones apropiadas resultan rápidos aunque
costosos procesos de neutralización. Pequeños volúmenes del agente son
requeridos, ya que estos neutralizantes son mucho más poderosos que la
caliza y la cal. Otra ventaja es que los productos de la reacción son solubles
y no aumentan las durezas a las aguas que se reciben. La soda cáustica
es mezclada normalmente en la parte de succión de la bomba de descarga
de los efluentes. Este método es apropiado para pequeños volúmenes, pero
para neutralizar grandes volúmenes de desecho acuoso, se necesitan especiales
equipos, de grandes dimensiones, como un depósito para el almacenamiento
del neutralizante con una bomba de diversas velocidades para la directa
adición del álcali a la corriente del desecho.
Discutimos hasta ahora cuatro métodos
para neutralización, sería conveniente el estudio de los costos de la neutralización
de desechos y de los agentes que hemos considerados.
Ya que el factor de basicidad (ver
tabla adjunta), es una de los factores vitales en la selección de un neutralizante,
se nos proviene no sólo de un cálculo, sino también un monograma para calcular
la cantidad de agente neutralizante requerido por galón de desecho. Se determina
así el valor de ácido por titilación de 5 ml de ácido sulfúrico con exceso
de hidróxido de sodio 0,5 N y una contratitulación con ácido clorhídrico
0,5 N donde el punto final de la titulación lo marca el viraje de color de
la fenolftaleína. El factor de basicidad de la cal ( o agente neutralizador)
es determinado por la titulación de 1 mg de muestra de agente alcalino con
exceso de ácido clorhídrico 0,5 N, hirviendo la muestra por quince minutos,
y contratitulando con hidróxido de sodio, donde el punto final de la titulación
lo da el viraje de color de la fenolafteína. El valor del ácido y del factor
de neutralización son luego conectados en el monograma de
HOAK para
encontrar la cantidad de agente requerido por galón de desecho ácido.
Cuando el hidróxido de sodio es usado
como neutralizante para el ácido sulfúrico, las siguientes ecuaciones tienen
lugar:
NaOH + H
2SO
4 NaHSO
4 +
H
2O,
desecho ácido
NaHSO
4 +
NaOH
Na
2SO
4 +H
2O.
La neutralización se produce en dos
pasos y los productos finales dependen del valor requerido para el pH final.
Por ejemplo, un tratamiento necesita de un pH final de 6, y el NaHSO
4 formaría
la mayor parte del producto; otro tratamiento podría requerir un pH de 8,
con lo cual la mayor parte del producto será Na
2SO
4.
Efluentes gaseosos
Control de los efluentes gaseosos.
Una norma sancionada por la experiencia es que
toda materia prima debe conocerse a fondo antes de utilizarla. por tanto,
habrá que aplicar este criterio a la contaminación, ya que la materia prima
que la provoca son los productos de combustión que, por tanto, se deben conocer
perfectamente.
Se vio que uno de los factores o parámetros que
inciden sobre la contaminación gaseosa detectable en un punto cualquiera
es la sobrelevación del penacho de humos, que a su vez depende de las energías
térmicas y cinética que se le comunique. Por tanto, es preciso determinar
la velocidad y temperaturas de los humos.
Como por otro lado se sabe que en el combustible
una cierta cantidad de azufre, como impureza, aunque también la proporción
del mismo es muy variable, será preciso determinar las cantidades reales
de anhídrido sulfúrico y sulfuroso emitidas.
Aunque la eficacia de los precipitadores electrostáticos
y mecánicos utilizados en la eliminación de polvo de los gases de combustión
es de sobra conocida, también se sabe que la resistividad y abrasividad de
las cenizas volantes es variable, como consecuencia de las diversas procedencias
del carbón que alimenta a la central, el rendimiento de los precititadores
varia. En consecuencia, otro dato que hay que determinar en el efluente será el
contenido de polvo de los gases de combustión y el rendimiento de la precipitación.
Por ultimo, si la combustión de la caldera no
tiene la eficacia deseable, probable que se alcance un valor de la escala
Ringelmann superior a 2, lo que plantea la necesidad de medir la densidad óptica
de los humos emitidos.
A continuación se examinaran los diversos sistemas
de control utilizados, en los que únicamente se estudia el principio físico
en que se basan pues se prescinde de cualquier tipo concreto de aparato.
El control continuo de la opacidad de los gases
de la combustión se efectúa de la forma siguiente: en un tramo recto de la
tubería de conducción de humos, ver figura, se coloca el sistema fotoeléctrico
de control, que consta, por una parte, de un dispositivo de proyección luminosa
de intensidad constante y, por otra, y exactamente en frente de esta, el
sistema receptor, constituido por una célula fotoeléctrica que mide la intensidad
luminosa recibida y a continuación la registra en un dispositivo de control.
Si se considerase deseable, se puede acoplar
a este equipo una unidad de control remoto y un sistema de alarma calibrado
de acuerdo con la legislación vigente o bien de tal modo que permita determinar
la eficacia de la combustión, pues cuanto mayor sea la intensidad luminosa
tanto mas eficaz será la combustión.
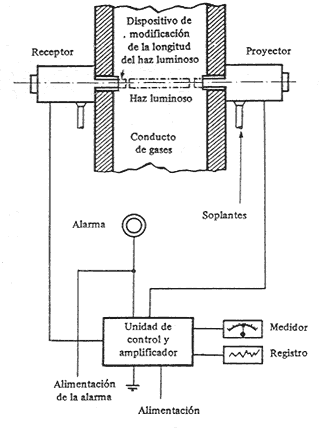
Esquema
de dispositivo fotoeléctrico
de medida de opacidad de los gases
Por regla general, los sistemas emisor y receptor
de estos equipos van provistos de unos dispositivos que tienen acortar o
alargar el recorrido del haz luminoso por el humo, si ello es preciso. Además,
para evitar los atascos producidos por la acumulación de polvo y hollín en
la conducción del equipo, el aparato dispone de un aparato de limpieza automática.
Con estos sistemas de medida el encargado de
la caldera esta siempre al corriente de la marcha de la combustión y, por
lo tanto, cuando sea preciso, puede adoptar las medidas necesarias para que
aquella pueda mantenerse en todo momento en condiciones optimas de régimen.
Por regla general, aun cuando funcionen bien,
las centrales térmicas emiten humos de opacidad superior a la normal durante
periodos cortos de tiempo que se corresponden con el encendido a partir de
la caldera fría hasta alcanzar el mínimo técnico de carga.
Determinación del dióxido de azufre.
El método de valoración mas usado para este compuesto
es el que utiliza iodo como reactivo. Para ello se prepara una disolución del
elemento de normalidad adecuada, (generalmente N/100) que se valora convenientemente.
Un pequeño volumen de esta disolución (10-40
ml) se coloca en un frasco lavador con placa perforada, adicionándole almidón
como indicador.
Se disponen adecuadamente los aparatos medidores
de temperatura y presión, tanto en el conducto efluente, como en el conducto
analizador, así como un medidor se caudal, según se indico con anterioridad.
La corriente gaseosa se hace pasar a través de
un frasco lavador, interrumpiéndose la operación en el momento justo en que
la coloración del reactivo absorbente desaparece.
Conocida la concentración de iodo en la disolución,
unos sencillos cálculos permiten determinar cuantitativamente el dióxido
de azufre que ha reaccionado en el frasco lavador. El volumen de corriente
gaseosa se corrige si es necesario en función de la presión y temperatura
medidos, y los resultados pueden expresarse en p.p.m. o bien en porcentaje.
Compuestos de azufre
El azufre es un elemento que se encuentra presente
en diversas proporciones en gran parte de los combustibles. Durante el proceso
de combustión se combina con el oxigeno para formar los correspondientes óxidos,
de os cuales los mas importantes son el dióxido y el trióxido.
Las cantidades de estos compuestos que se vierten
a la atmósfera son extraordinariamente elevadas, alcanzando cifras de millones
de toneladas al año.
A temperatura ambiente, el dióxido de azufre
es un gas que condensa con facilidad. Es incoloro, de olor picante e irritante
y más pesado que el aire, poseyendo un elevado poder de corrosión. Químicamente
puede actuar como oxidaste (por ejemplo frente al sulfuro de hidrogeno),
o como reductor (con los halógenos).
El trióxido de azufre es un liquido incoloro
de elevada afinidad por el agua, con la que forma ácido sulfúrico, y de propiedades
oxidasteis. La aparición de este compuesto en la atmósfera se puede deber
a tres causas:
- Oxidación directa del SO2
- Oxidación catalítica del SO2
- Descomposición de los sulfatos presentes en
los combustibles
De las tres formas posibles, parece ser la segunda
la que interviene en mayor escala, actuando como catalizadores diversos compuestos
metálicos existentes en las porciones inorgánicas de los combustibles.
La concentración de dióxido de azufre en la atmósfera
presenta variaciones típicamente estacionales debido al mayor empleo de combustibles
las épocas frías del invierno.
Efecto sobre las plantas.
La contaminación atmosférica afecta a los vegetales
de diferentes maneras. A grandes rasgos los dados ocasionados pueden agruparse
en los tres siguientes grupos:
- Afecciones de los tejidos de las hojas con
necrosis parciales.
- Clorosis y otros cambios de color.
- Alteraciones en el crecimiento.
Estas afecciones obedecen a dos causas diferentes:
- Penetración directa de los contaminantes en
los vegetales a través de los estomas de las hojas
- Deposición sobre las diferentes partes de la
planta, influyendo e la fotosíntesis.
Las afecciones en los tejidos de las hojas provocan
la plasmolisis de las células vegetales que puede ser parcial si el causante
es el nitrato de peracetilio (PAN) o el ozono, o total en los compuestos
fluorados y del dióxido de azufre. Las hojas afectadas presentan primeramente
un suave aspecto de estar empapadas en agua y algunas magulladuras pequeñas.
La clorolisis es la perdida de clorofila, pigmento
natural de las plantas, y puede se comparada con la anemia de los animales.
La desaparición de la clorofila da lugar a nuevos colores a partir de otros
pigmentos presentes. Por regla general, la coloración es característica del
contaminante que esta causando el fenómeno. Así, por ejemplo, una coloración
blanquecina es producida por el dióxido de azufre y una marrón por el flúor.
Lógicamente, las afecciones de las hojas de los
vegetales, donde se produce la transformación de numerosas sustancias inorgánicas
en otras nutritivas para la planta, puede afectar seriamente el crecimiento
de ella.
En la práctica, han sido observadas numerosas
deficiencias con ciertas plantas situadas en ambientes muy contaminados,
tales como, perdida precoz de las hojas, disminución del tamaño del fruto,
crecimiento inferior al normal y prematuro envejecimiento general.
El crecimiento de la planta, concretamente, se
ve seriamente afectado por la contaminación atmosférica, pues esta tiene
una influencia muy fuerte sobre la auxina, hormona vegetal muy importante
en la regulación de la evolución general de la planta, y cuya acción es afectada
por cantidades de contaminantes sumamente débiles.
Dióxido de azufre
Las lesiones que produce en las plantas son normalmente
de origen local. Las células admiten cierta concentración máxima del compuesto,
que cuando es superada provoca el fin de su actividad y la muerte posterior.
Esta toxicidad se atribuye generalmente a las
propiedades reductoras del dióxido, y la coloración característica que comunica
a las zonas afectadas es de tonos amarillentos y rojizos.
La sensibilidad de las diferentes plantas a este
contaminante es muy variedad. Experimentalmente ha podido ser determinada
la resistencia relativa de numerosas especies y que se atribuye a la alfalfa
el valor unidad.